May 09, 2023PRESS RELEASE
Team succeeds in elucidating a new mechanism in causal gene mutations found in early-onset Parkinson’s disease
~ Mechanism involves removal of damaged mitochondria facilitated by translocation-mediated protein ~
Keyword:RESEARCH
OBJECTIVE.
A team of scientists that includes Professor Toshihiko Oka of Rikkyo University’s College of Science, has succeeded in elucidiating a novel mechanism caused by gene mutations responsible for early onset Parkinson’s disease (PD). The results of this study will be published online in the U.S. journal “Cell Reports” at midnight on May 9, 2023 (Japan time, 11:00 a.m., U.S. EST).
1) Outline of research results
Mitochondria, which have received a great deal of attention in recent years, are organelles (structures surrounded by membranes) found in cells that generate energy for use in the human body. Mitochondria are sometimes damaged by reactive oxygen species*1 generated when a large amount of energy is produced. The accumulation of such damage compromises mitochondrial functions. Cells, on the other hand, actively remove and degrade damaged mitochondria to maintain efficient energy production, in what is known as “mitochondrial quality control*2.”
PINK1, a gene product that causes early-onset PD, is a protein that functions as a sensor of damaged mitochondria. PINK1 is activated on damaged mitochondria and then facilitates their removal. One factor that triggers PD is believed to be insufficient removal of damaged mitochondria. Among PINK1 mutantions found in PD patients, only some are directly involved in its enzymatic activity. It is unclear how the remaining mutations in PINK1 affect the functions.
In the present study, Oka’s research team successfully discovered a new mechanism underlying PINK1 activation on damaged mitochondria and PINK1 mutations that inhibit the mechanism. The study was conducted in collaboration with groups including those led by Professor Toshiya Endo of the Faculty of Life Sciences at Kyoto Sangyo University, and Professor Hidetaka Kosako of the Fujii Memorial Institute of Medical Sciences at Tokushima University.
In the study, researchers including Shiori Akabane, who is affiliated with both Rikkyo University and Kyoto Sangyo University, found that PINK1and TIM23, a protein mediating protein translocation into mitochondria, form a protein complex. They also discovered that TIM23 safeguards PINK1 against proteases, protein-degrading enzymes, in damaged mitochondria, thereby facilitating the removal of damaged mitochondria. In addition, they found some pathogenic PINK1 mutations impair interactions between PINK1 and TIM23, thereby inhibiting the activation of PINK1.
The research results are expected to deepen the understanding of the mechanism underlying elimination of damaged mitochondria. The findings are also expected to help develop a better PD treatment.
We are pleased to announce that the research results will be published online in the U.S. journal “Cell Reports” dated May 9, 2023 (Japan time).
PINK1, a gene product that causes early-onset PD, is a protein that functions as a sensor of damaged mitochondria. PINK1 is activated on damaged mitochondria and then facilitates their removal. One factor that triggers PD is believed to be insufficient removal of damaged mitochondria. Among PINK1 mutantions found in PD patients, only some are directly involved in its enzymatic activity. It is unclear how the remaining mutations in PINK1 affect the functions.
In the present study, Oka’s research team successfully discovered a new mechanism underlying PINK1 activation on damaged mitochondria and PINK1 mutations that inhibit the mechanism. The study was conducted in collaboration with groups including those led by Professor Toshiya Endo of the Faculty of Life Sciences at Kyoto Sangyo University, and Professor Hidetaka Kosako of the Fujii Memorial Institute of Medical Sciences at Tokushima University.
In the study, researchers including Shiori Akabane, who is affiliated with both Rikkyo University and Kyoto Sangyo University, found that PINK1and TIM23, a protein mediating protein translocation into mitochondria, form a protein complex. They also discovered that TIM23 safeguards PINK1 against proteases, protein-degrading enzymes, in damaged mitochondria, thereby facilitating the removal of damaged mitochondria. In addition, they found some pathogenic PINK1 mutations impair interactions between PINK1 and TIM23, thereby inhibiting the activation of PINK1.
The research results are expected to deepen the understanding of the mechanism underlying elimination of damaged mitochondria. The findings are also expected to help develop a better PD treatment.
We are pleased to announce that the research results will be published online in the U.S. journal “Cell Reports” dated May 9, 2023 (Japan time).
2) Research Background
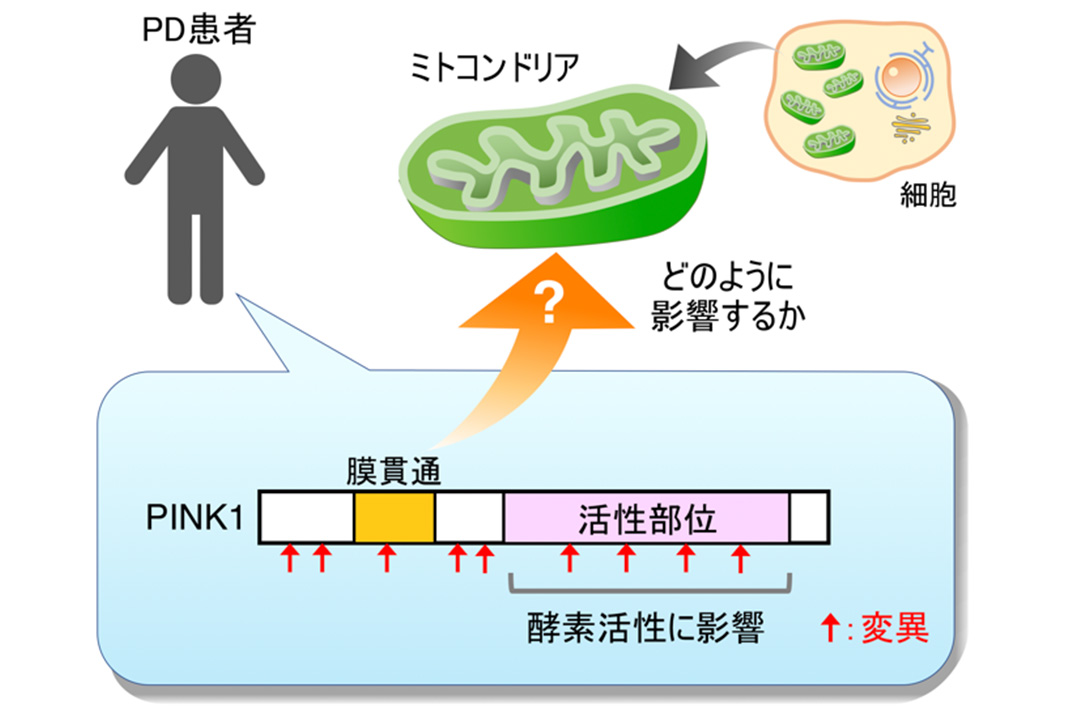
Fig. 1: PINK1 mutations found in PD patients
PINK1 protein, a mitochondrial sensor, is delivered to mitochondria after being synthesized in cytosol. In normal, undamaged mitochondria, PINK1 is degraded soon, leading to no PINK1 accumulation. But in damaged mitochondria, PINK1 is stabilized by resistance to the degradation, which results in the formation of protein complexes. This leads to increased PINK1 enzymatic activity, followed by the recruitment of Parkin, another gene product that causes PD, to damaged mitochondria. Parkin adds a protein that serves as a label for mitochondrial degradation to damaged mitochondria, thus facilitating active removal of mitochondria.
In a nutshell, the accumulation of PINK1 and formation of protein complexes play first key steps in actively removing damaged mitochondria. However, it was not fully clear with which PINK1 protein forms protein complexes. Furthermore, none of the PINK1 mutations found in PD patients was known to inhibit PINK1 accumulation and protein complex formation, excluding those affecting enzymatic activity (See Fig. 1)
In a nutshell, the accumulation of PINK1 and formation of protein complexes play first key steps in actively removing damaged mitochondria. However, it was not fully clear with which PINK1 protein forms protein complexes. Furthermore, none of the PINK1 mutations found in PD patients was known to inhibit PINK1 accumulation and protein complex formation, excluding those affecting enzymatic activity (See Fig. 1)
3) Content and results of research
Akabane and her colleagues, in collaboration with researchers from the Fujii Memorial Institute of Medical Sciences at Tokushima University, discovered TIM23 after conducting an exhaustive analysis of proteins that interact with PINK1 in damaged mitochondria. TIM23 is one of the proteins comprising the translocation machinery: a pore in the mitochondrial inner membranes through which proteins are transported into mitochondria. Since proteins are not transported into damaged mitochondria, the analysis of TIM23 functions was previously conducted using only normal mitochondria. In the present study, however, the team used damaged mitochondria with reduced TIM23 protein levels. They found that PINK1 accumulation was strongly inhibited and that subsequent Parkin’s mitochondrial targeting was impaired. The results therefore show that TIM23 has two functions: TIM23 serves as a protein translocation machinery*3 in normal mitochondria and facilitates PINK1 activation in damaged mitochondria.
The team then investigated which protease caused reduction in PINK1 protein levels in the absence of TIM23. They confirmed that the PINK1 protein levels were restored by silencing the expression of OMA1, the inner mitochondrial membrane protease. OMA1 is known to regulate mitochondrial morphology and stress response upon activation in damaged mitochondria. Since PINK1 is also a substrate, it is assumed that PINK1 is degraded by OMA1 in the absence of TIM23.
The above results demonstrate TIM23 facilitates PINK1 activation in damaged mitochondria by safeguarding against OMA1-mediated degradation. TIM23’s safeguarding mechanism has not only a new function of the mitochondrial translocator, but also a new role of TIM23 in PINK1 activation.
The team then investigated which protease caused reduction in PINK1 protein levels in the absence of TIM23. They confirmed that the PINK1 protein levels were restored by silencing the expression of OMA1, the inner mitochondrial membrane protease. OMA1 is known to regulate mitochondrial morphology and stress response upon activation in damaged mitochondria. Since PINK1 is also a substrate, it is assumed that PINK1 is degraded by OMA1 in the absence of TIM23.
The above results demonstrate TIM23 facilitates PINK1 activation in damaged mitochondria by safeguarding against OMA1-mediated degradation. TIM23’s safeguarding mechanism has not only a new function of the mitochondrial translocator, but also a new role of TIM23 in PINK1 activation.
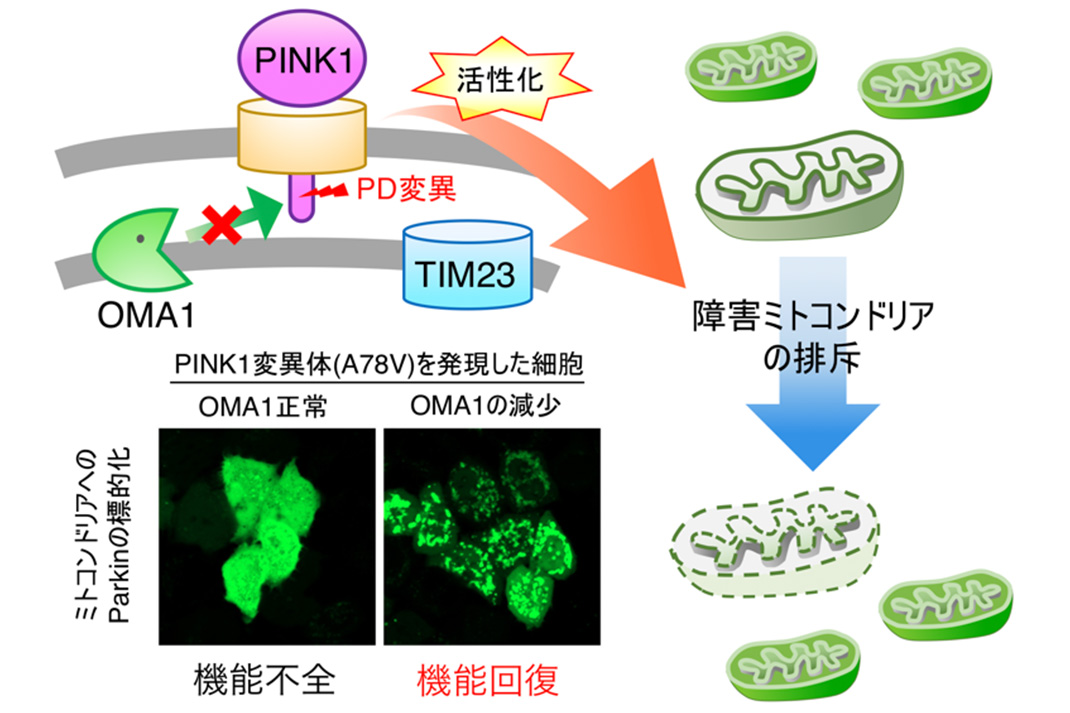
Fig. 2: OMA1 inactivation facilitates the removal of damaged mitochondria by suppressing PINK1 mutations
The team also looked into whether PINK1 mutations found in PD patients affect the safeguarding mechanism mediated by TIM23. It used 24 kinds of PINK1 mutations. Three of them were confirmed to weaken interaction between PINK1 and TIM23.
These PINK1 variants can be activated by reducing OMA1 levels, thereby leading to efficient mitochondrial targeting of Parkin protein. This result suggests that reduced OMA1 levels facilitate the removal of damaged mitochondria by triggering the accumulation and activation of PINK1 pathogenic variants. This points to the possibility of recovering from damage caused by PINK1 mutations (See Fig. 2).
These PINK1 variants can be activated by reducing OMA1 levels, thereby leading to efficient mitochondrial targeting of Parkin protein. This result suggests that reduced OMA1 levels facilitate the removal of damaged mitochondria by triggering the accumulation and activation of PINK1 pathogenic variants. This points to the possibility of recovering from damage caused by PINK1 mutations (See Fig. 2).
4) Future prospects
The results of the present research suggest that a new molecular mechanism in certain PINK1 mutations leads to the outbreak of PD, in which PINK1 (mitochondrial sensor) protein levels are reduced due to impaired interactions between PINK1 and TIM23. This is expected to help launch a new PD treatment strategy by developing drugs that can inhibit OMA1 activation and strengthen interactions between PINK1 and TIM23.
Glossary
*1 Reactive oxygen species (ROS):
Chemical compounds that contain highly reactive oxygen molecules. A typical ROS is hydrogen peroxide.
*2 Mitochondrial quality control:
A mechanism to maintain and control the quality of mitochondria by selectively degrading and removing damaged mitochondria with reduced functions to raise the ratio of energized mitochondria.
*3 Protein translocation machinery:
Protein complex consisting of a pore used to translocate proteins across organellar membranes. Many of the proteins constituting organelles are synthesized in cytosol, after which are translocated across membranes of target organelles.
Chemical compounds that contain highly reactive oxygen molecules. A typical ROS is hydrogen peroxide.
*2 Mitochondrial quality control:
A mechanism to maintain and control the quality of mitochondria by selectively degrading and removing damaged mitochondria with reduced functions to raise the ratio of energized mitochondria.
*3 Protein translocation machinery:
Protein complex consisting of a pore used to translocate proteins across organellar membranes. Many of the proteins constituting organelles are synthesized in cytosol, after which are translocated across membranes of target organelles.
Article information
- Title: TIM23 facilitates PINK1 activation by safeguarding against OMA1-mediated degradation in damaged mitochondria
- Authors: Shiori Akabane, Kiyona Watanabe, Hidetaka Kosako, Shun-ichi Yamashita, Kohei Nishino, Masahiro Kato, Shiori Sekine, Tomotake Kanki, Noriyuki Matsuda, Toshiya Endo and Toshihiko Oka
- Journal: Cell Reports
- Link: https://www.cell.com/cell-reports/home